Retinoblastoma is an uncommon eye cancer impacting about 300 children each year. The disease occurs most often in children under the age of 4 years, with 80% of cases taking place under age 3 years and virtually none above age 6 years. Many children (99%) with this disease will be cured.
Childhood retinoblastoma originates in a part of the eye called the retina. The retina is a thin layer of nerve tissue that coats the back of the eye and permits a person to see. Retinoblasts (immature cells of the retina) multiply during pregnancy and early life to make sufficient cells to create the retina. Retinoblastoma is a cancer that impacts these retinoblasts, causing them to increase too much, and not to mature fully (a process called distinction). In children this can take place in one eye (unilateral) or both eyes (bilateral).
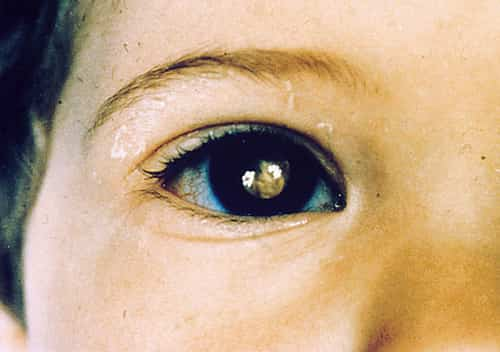
Symptoms of Retinoblastoma
Doctors can determine retinoblastoma on a routine well-baby or well-child exam. Parents likewise discover symptoms such as:
- A pupil that looks white or black rather of red when light hits it
- A crossed eye (looking either towards the nose or toward the ear).
- Poor vision.
- A red, painful eye.
- A bigger pupil.
Types of Retinoblastoma
There are two kinds of the disease, a heritable kind and non-heritable form (all cancers are considered hereditary because anomalies of the genome are required for their advancement, but this does not suggest that they are heritable, or sent to offspring). Approximately 55% of children with retinoblastoma have the non-heritable kind. If there is no history of the disease within the family, the disease is labeled “sporadic”, however this does not always show that it is the non-heritable kind. Bilateral retinoblastomas are typically heritable, while unilateral retinoblastomas are commonly non-heritable.
In about two-thirds of cases, only one eye is impacted (unilateral retinoblastoma); in the other third, tumors develop in both eyes (bilateral retinoblastoma). The number and size of tumours on each eye may differ. In certain cases, the pineal gland or the suprasellar or parasellar area (or in really rare cases other midline intracranial places) is likewise affected (trilateral retinoblastoma). The position, size and quantity of tumours are thought about when picking the kind of treatment for the disease.
What Causes Retinoblastoma?
Anomaly of genes, found in chromosomes, can affect the way in which cells grow and develop within the body. Alterations in RB1 or MYCN can trigger retinoblastoma.
RB1
In children with the heritable hereditary form of retinoblastoma there is an anomaly in the RB1 gene on chromosome 13. RB1 was the first tumor suppressor gene cloned. Although RB1 engages with over 100 cell proteins, its negative regulator impact on the cell cycle primarily occurs from binding and inactivation of the transcription element E2F, hence repressing the transcription of genes which are needed for the S stage.
The faulty RB1 gene can be acquired from either parent; in some children, nevertheless, the anomaly occurs in the early stages of fetal development. The expression of the RB1 allele is autosomal dominant with 90% penetrance.
Acquired types of retinoblastomas are more likely to be bilateral. In addition, acquired uni- or bilateral retinoblastomas may be related to pineoblastoma and other deadly midline supratentorial primitive neuroectodermal tumors (PNET) with a miserable outcome; retinoblastoma concurrent with a PNET is known as trilateral retinoblastoma. A recent meta-analysis has actually revealed that survival of trilateral retinoblastoma has increased significantly over the last decades.
The development of retinoblastoma can be explained by the two-hit model. Inning accordance with the two-hit design, both alleles have to be affected, so two events are needed for the retinal cell or cells to become tumors. The first mutational occasion can be inherited (germline or constitutional) which will then be present in all cells in the body. The 2nd “hit” leads to the loss of the remaining normal allele (gene) and happens within a specific retinal cell. In the erratic, nonheritable form of retinoblastoma, both mutational events take place within a single retinal cell after fertilization (somatic occasions); sporadic retinoblastoma has the tendency to be unilateral.
Numerous approaches have actually been developed to discover the RB1 gene mutations. Efforts to correlate gene mutations to the stage at presentation have actually not shown convincing evidence of a connection.
MYCN
Somatic amplification of the MYCN oncogene is responsible for some cases of non-hereditary, early-onset, aggressive, unilateral retinoblastoma. Although MYCN amplification accounted for only 1.4% of retinoblastoma cases, researchers identified it in 18% of babies identified at less than 6 months of age. Mean age at diagnosis for MYCN retinoblastoma was 4.5 months, compared with 24 months for those who had non-familial unilateral disease with two RB1 gene anomalies.
Diagnosis
Screening for retinoblastoma ought to be part of a “well baby” screening for newborns during the first three months of life, to consist of:
- The red reflex: looking for a normal reddish-orange reflection from the eye’s retina with an ophthalmoscope or retinoscope from roughly 30 cm/ 1 foot, typically done in a dimly lit or dark space.
- The corneal light reflex/ Hirschberg test: looking for in proportion reflection of beam in the very same spot on each eye when a light is shined into each cornea, to assist determine whether the eyes are crossed.
- Eye assessment: looking for any structural abnormalities.
- Bryan Shaw assisted develop a smart-phone app that can spot leukocoria in images.
Treatment of Retinoblastoma in Children
The concern of retinoblastoma treatment is to maintain the life of the child, then to preserve vision, then to lessen complications or side effects of treatment. The specific course of treatment will depend upon the individual case and will be chosen by the eye doctor in conversation with the paediatric oncologist. Children with participation of both eyes at medical diagnosis usually require multimodality therapy (chemotherapy, local therapies).
The various treatment modalities for retinoblastoma consists of:
- Enucleation of the eye. Most patients with unilateral disease present with sophisticated intraocular disease and for that reason typically undergo enucleation, which leads to a cure rate of 95%. In bilateral Rb, enucleation is usually reserved for eyes that have failed all known reliable treatments or without helpful vision.
- External beam radiotherapy (EBR). The most common sign for EBR is for the eye in a young child with bilateral retinoblastoma who has active or frequent disease after conclusion of chemotherapy and local therapies. Nevertheless, patients with genetic disease who got EBR therapy are reported to have a 35% risk of 2nd cancers.
- Brachytherapy. Brachytherapy includes the placement of a radioactive implant (plaque), generally on the sclera adjacent to the base of a tumor. It used as the main treatment or, more frequently, in patients with small tumors or in those who had stopped working preliminary therapy including previous EBR therapy.
- Thermotherapy. Thermotherapy involves the application of heat straight to the tumor, typically in the form of infrared radiation. It is also used for little tumors.
- Laser photocoagulation. Laser photocoagulation is suggested just for small posterior tumors. An argon or diode laser or a xenon arc is used to coagulate all the blood supply to the tumor.
- Cryotherapy. Cryotherapy causes damage to the vascular endothelium with secondary thrombosis and infarction of the tumor tissue by rapidly freezing it. Cryotherapy may be used as primary therapy for small peripheral tumors or for small persistent tumors previously treated with other approaches.
- Systemic chemotherapy. Systemic chemotherapy has become forefront of treatment in the past decade, in the search of globe protecting measures and to avoid the adverse effects of EBR therapy. The common indicators for chemotherapy for intraocular retinoblastoma include tumors that are large which can not be treated with local treatments alone in children with bilateral tumors. It is also used in patients with unilateral disease when the tumors are little but can not be managed with local treatments alone.
- Intra-arterial chemotherapy. Chemotherapeutic drugs are administered in your area by means of a thin catheter threaded through the groin, through the aorta and the neck, directly into the optic vessels.
- Nano-particulate chemotherapy. To reduce the adverse results of systemic therapy, subconjuctival (local) injection of nanoparticle carriers consisting of chemotherapeutic agents (carboplatin) has been developed which has revealed appealing lead to the treatment of retinoblastoma in animal designs without adverse results.
- Chemoreduction. A combined technique using chemotherapy to initially decrease the size of the tumor, and adjuvant focal treatments, such as transpupillary thermotherapy, to control the tumor.
What to Expect?
In the industrialized world, retinoblastoma has one of the best cure rates of all childhood cancers (95-98%), with more than 9 out of every 10 victims surviving into the adult years. In the UK, around 40 to 50 new cases are diagnosed each year. Good prognosis depends upon early discussion of the child in health center. Late discussion of the child in health center is associated with poor prognosis. Survivors of genetic retinoblastoma have a greater risk of establishing other cancers later on in life.