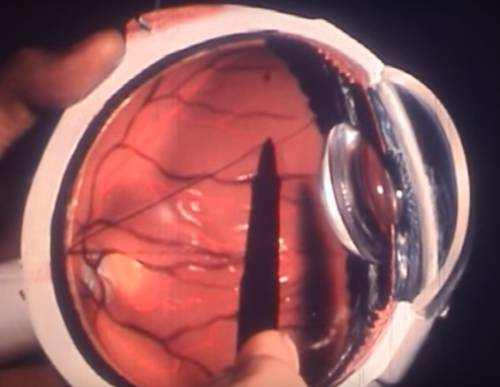
Retinitis pigmentosa (RP) is the name given to a group of inherited eye diseases that affect the retina (the light-sensitive part of the eye). RP causes the breakdown of photoreceptor cells (cells in the retina that detect light). Photoreceptor cells capture and process light helping us to see. As these cells breakdown and die, patients experience progressive vision loss.
The most typical function of all kinds of RP is a gradual breakdown of rods (retinal cells that identify dim light) and cones (retinal cells that find light and color). The majority of forms of RP first cause the breakdown of rod cells. These types of RP, often called rod-cone dystrophy, typically begin with night blindness. Night blindness is somewhat like the experience normally spotted people experience when entering a dark film theatre on a bright, sunny day. Nevertheless, patients with RP can not change well to dark and dimly lit environments.
Vision Support: Quality AREDS 2 Formula eye vitamins can be found on Amazon.
What Are the Symptoms of Retinitis Pigmentosa?
As the disease advances and more rod cells breakdown, patients lose their peripheral vision (one-track mind). People with RP typically experience a ring of vision loss in their periphery, however maintain clear central vision. Others report the feeling of tunnel vision, as though they see the world through a straw. Many patients with retinitis pigmentosa keep a small degree of central vision throughout their life.
Other types of RP, in some cases called cone-rod dystrophy, first affect central vision. Patients first experience a loss of main vision that can not be fixed with glasses or contact lenses. With the loss of cone cells likewise comes disruptions in color perception. As the disease advances, rod cells degenerate causing night blindness and peripheral vision.
Symptoms of RP are most often acknowledged in children, teenagers and young people, with progression of the disease continuing throughout the person’s life. The pattern and degree of visual loss vary.
What Causes Retinitis Pigmentosa?
Retinitis pigmentosa is an inherited disorder, and for that reason not brought on by injury, infection or other external or environmental aspects. People struggling with RP are born with the disorder currently programmed into their cells. Medical professionals can see the first signs of retinitis pigmentosa in affected children as early as age 10. Research suggests that several different types of gene anomalies (changes in genes) can send faulty messages to the retinal cells which leads to their progressive degeneration. In most cases, the disorder is linked to a recessive gene, a gene that should be inherited from both parents in order to cause the disease.
Expert Choice: Recommended by eye care professionals for managing dry eyes, styes, and blepharitis, this Heated Eye Mask provides the consistent moist heat therapy endorsed by the American Academy of Ophthalmology. It’s an effective, reusable solution for soothing irritation and improving oil gland function. Available on Amazon.
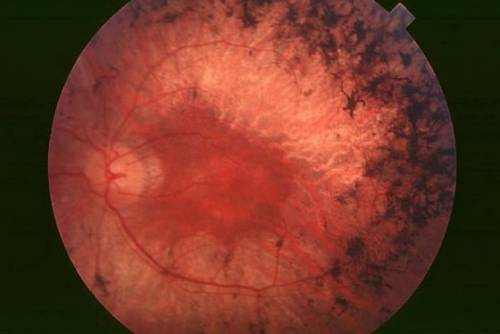
However dominant genes and genes on the X chromosome likewise have actually been linked to retinitis pigmentosa. In these cases, just one parent has passed the disease gene. In some cases, a new mutation causes the disease to take place in a person who does not have a family history of the disease. The disorder also can show up as part of other syndromes, such as Bassen-Kornzweig disease or Kearns-Sayre syndrome.
Treatment for Retinitis Pigmentosa
There is no known remedy for retinitis pigmentosa. Nevertheless, there are few treatment choices such as light avoidance and/or the use of low-vision helps to slow down the progression of RP. Some professionals also think about vitamin A as a possible treatment alternative to decrease the progression of RP. Research suggests taking high dosages of vitamin A (15,000 IU/day) may slow progression a little in some people, however the outcomes are not strong. Taking excessive vitamin A can be poisonous and the effects of vitamin A on the disease is relatively weak. More research should be carried out before this is an extensively accepted type of therapy.
Research is also being conducted in areas such as gene therapy research, transplant research, and retinal prosthesis. Given that RP is usually the result of a faulty gene, gene therapy has ended up being an extensively checked out area for future research. The objective of such research would be to discover ways healthy genes can be placed into the retina. Attempts at transplanting healthy retinal cells into sick retinas are being made experimentally and have actually not yet been thought about as clinically safe and successful.
Specialized Care: For persistent itching or flaking, eye care specialists recommend targeted formulas like this BetterLids Eyelid Ointment . This preservative-free, dermatologist-tested oat complex is a top choice for soothing sensitive skin and managing symptoms of blepharitis. Available for easy ordering on Amazon.
Retinal prosthesis is also a crucial area of expedition because the prosthesis, a man-made device planned to change a harmed body part, can be designed to take control of the function of the lost photoreceptors by electrically stimulating the staying healthy cells of the retina.Through electrical stimulation, the triggered ganglion cells can provide a visual signal to the brain. The visual scene caught by an electronic camera is sent by means of electro-magnetic radiation to a little decoder chip located on the retinal surface. Information and power are then sent out to a set of electrodes connected to the decoder. Electrical current death from private electrodes stimulate cells in the suitable areas of the retina representing the features in the visual scene.
What Do We Know About Heredity and Retinitis Pigmentosa?
Since RP is an inherited disorder, it can possibly impact another family member. With retinal cells being amongst the most specialized cells in the human body, they depend upon a variety of unique genes to develop vision. A disease-causing mutation in any among these genes can cause vision loss. Researchers have actually discovered over 100 genes that can include mutations leading to retinitis pigmentosa. Around 50 percent of RP cases are isolated and have no previous household history. The cause of these cases can not be discussed. Other cases of RP, where household history has actually been figured out, fall under 3 primary classifications: autosomal recessive, autosomal dominant, and X-linked recessive.
Autosomal recessive RP happens when both parents are untouched carriers of the very same defective gene. The opportunities of a child being impacted is one in 4. This suggests the afflicted child must acquire the faulty gene from each parent. The possibilities of a parent having an untouched child who would be a provider of the malfunctioning gene is one in two. The opportunity of parents having a child totally without the RP gene is one in four.
In autosomal dominant RP, the disease exists in males or women just when a single copy of the gene is defective. Usually, among the parents is impacted by the disease. The chance is one in two of any offered offspring being affected by the disease, if the affected parent has one normal and one faulty gene.
X-linked recessive RP might happen in offspring in two methods. The daddies can be affected or mothers can be providers of the malfunctioning gene. If the daddy is impacted, all children will be unaffected and all daughters will be providers. If the mother is the carrier, 1 in 2 kids will be impacted and 1 in 2 children will be carriers. In families with the X-linked type, just males are affected, while women carry the genetic quality but do not experience major vision loss.